
KAMENOVA Stefaniya
- CEES, University of Oslo, Oslo, Norway
- Biodiversity, Climate change, Food webs, Herbivory, Molecular ecology, Terrestrial ecology
- recommender
Recommendation: 1
Reviews: 0
Recommendation: 1
A flexible pipeline combining clustering and correction tools for prokaryotic and eukaryotic metabarcoding
A flexible pipeline combining clustering and correction tools for prokaryotic and eukaryotic metabarcoding
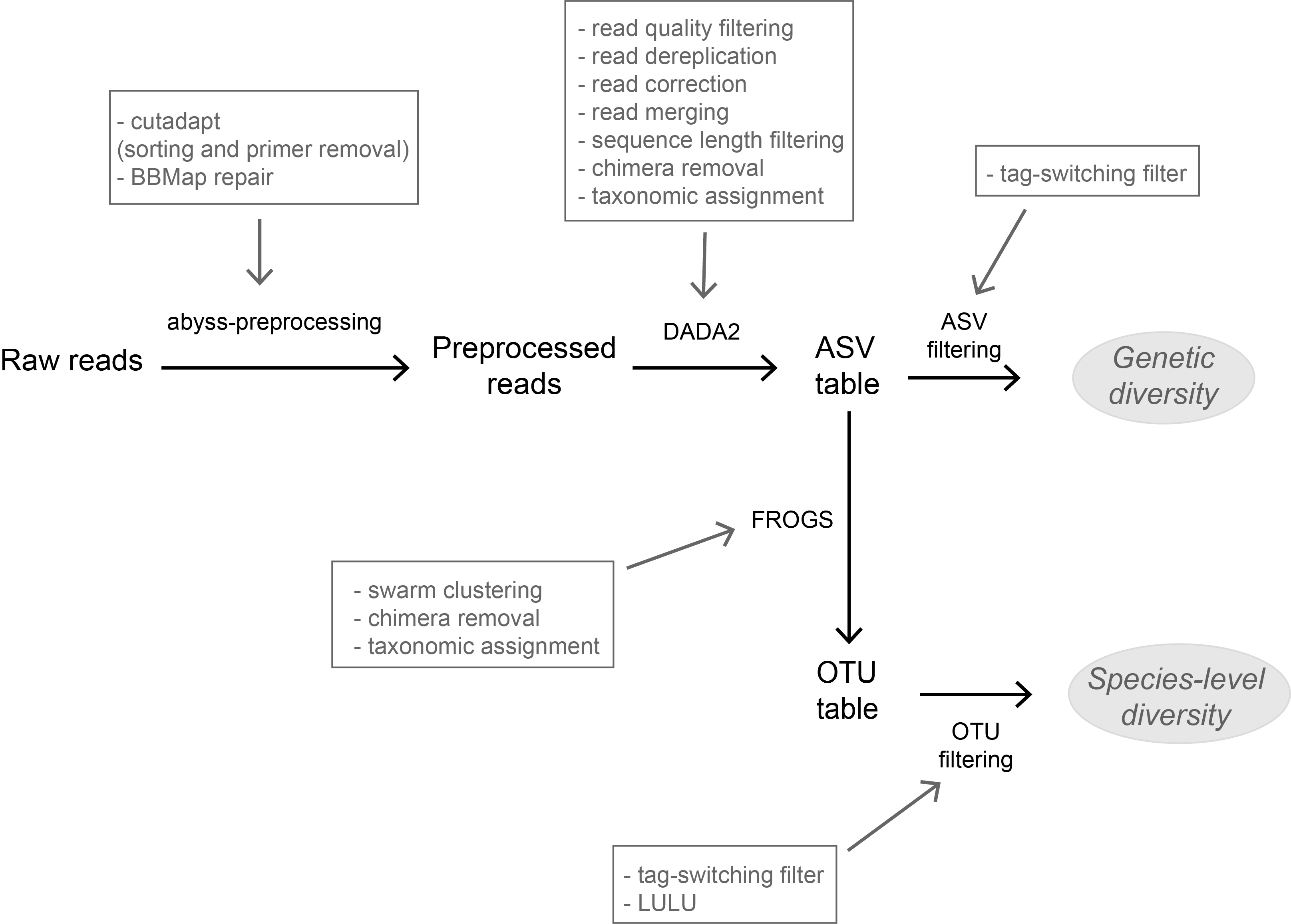
Recommended by Stefaniya Kamenova based on reviews by Tiago Pereira and 1 anonymous reviewerHigh-throughput sequencing-based techniques such as DNA metabarcoding are increasingly advocated as providing numerous benefits over morphology‐based identifications for biodiversity inventories and ecosystem biomonitoring [1]. These benefits are particularly apparent for highly-diversified and/or hardly accessible aquatic and marine environments, where simple water or sediment samples could already produce acceptably accurate biodiversity estimates based on the environmental DNA present in the samples [2,3]. However, sequence-based characterization of biodiversity comes with its own challenges. A major one resides in the capacity to disentangle true biological diversity (be it taxonomic or genetic) from artefactual diversity generated by sequence-errors accumulation during PCR and sequencing processes, or from the amplification of non-target genes (i.e. pseudo-genes). On one hand, the stringent elimination of sequence variants might lead to biodiversity underestimation through the removal of true species, or the clustering of closely-related ones. On the other hand, a more permissive sequence filtering bears the risks of biodiversity inflation. Recent studies have outlined an excellent methodological framework for addressing this issue by proposing bioinformatic tools that allow the amplicon-specific error-correction as alternative or as complement to the more arbitrary approach of clustering into Molecular Taxonomic Units (MOTUs) based on sequence dissimilarity [4,5]. But to date, the relevance of amplicon-specific error-correction tools has been demonstrated only for a limited set of taxonomic groups and gene markers.
The study of Brandt et al. [6] successfully builds upon existing methodological frameworks for filling this gap in current literature. By proposing a bioinformatic pipeline combining Amplicon Sequence Variants (ASV) curation with MOTU clustering and additional post-clustering curation, the authors show that contrary to previous recommendations, ASV-based curation alone does not represent an adequate approach for DNA metabarcoding-based inventories of metazoans. Metazoans indeed, do exhibit inherently higher intra-specific and intra-individual genetic variability, necessarily leading to biased biodiversity estimates unbalanced in favor of species with higher intraspecific diversity in the absence of MOTU clustering. Interestingly, the positive effect of additional clustering showed to be dependent on the target gene region. Additional clustering had proportionally higher effect on the more polymorphic mitochondrial COI region (as compared to the 18S ribosomal gene). Thus, the major advantage of the study lies in the provision of optimal curation parameters that reflect the best possible balance between minimizing the impact of PCR/sequencing errors and the loss of true biodiversity across markers with contrasting levels of intragenomic variation. This is important as combining multiple markers is increasingly considered for improving the taxonomic coverage and resolution of data in DNA metabarcoding studies.
Another critical aspect of the study is the taxonomic assignation of curated OTUs (which is also the case for the majority of DNA metabarcoding-based biodiversity assessments). Facing the double challenge of focusing on taxonomic groups that are both highly diverse and poorly represented in public sequence reference databases, the authors failed to obtain high-resolution taxonomic assignments for several of the most closely-related species. As a result, taxa with low divergence levels were clustered as single taxonomic units, subsequently leading to underestimation of true biodiversity present. This finding adds to the argument that in order to be successful, sequence-based techniques still require the availability of comprehensive, high-quality reference databases.
Perhaps the only regret we might have with the study is the absence of mock community validation for the prokaryotes compartment. Even though the analyses of natural samples seem to suggest a positive effect of the curation pipeline, the concept of intra- versus inter-species variation in naturally occurring prokaryote communities remains at best ambiguous. Of course, constituting a representative sample of taxonomically-resolved prokaryote taxa from deep-sea habitats does not come without difficulties but has the benefit of opening opportunities for further studies on the matter.
References
[1] Porter, T. M., and Hajibabaei, M. (2018). Scaling up: A guide to high-throughput genomic approaches for biodiversity analysis. Molecular Ecology, 27(2), 313–338. doi: 10.1111/mec.14478
[2] Valentini, A., Taberlet, P., Miaud, C., Civade, R., Herder, J., Thomsen, P. F., … Dejean, T. (2016). Next-generation monitoring of aquatic biodiversity using environmental DNA metabarcoding. Molecular Ecology, 25(4), 929–942. doi: 10.1111/mec.13428
[3] Leray, M., and Knowlton, N. (2015). DNA barcoding and metabarcoding of standardized samples reveal patterns of marine benthic diversity. Proceedings of the National Academy of Sciences, 112(7), 2076–2081. doi: 10.1073/pnas.1424997112
[4] Callahan, B. J., McMurdie, P. J., and Holmes, S. P. (2017). Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. The ISME Journal, 11(12), 2639–2643. doi: 10.1038/ismej.2017.119
[5] Edgar, R. C. (2016). UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing. BioRxiv, 081257. doi: 10.1101/081257
[6] Brandt, M. I., Trouche, B., Quintric, L., Wincker, P., Poulain, J., and Arnaud-Haond, S. (2020). A flexible pipeline combining clustering and correction tools for prokaryotic and eukaryotic metabarcoding. BioRxiv, 717355, ver. 3 peer-reviewed and recommended by PCI Ecology. doi: 10.1101/717355